
All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Lissencephaly of the brain
Medical expert of the article

Last reviewed: 12.07.2025
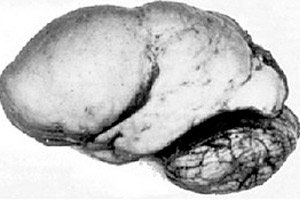
Among organic cerebral pathologies, such a congenital anomaly of brain development as lissencephaly stands out, the essence of which lies in the almost smooth surface of the cortex of its hemispheres - with an insufficient number of convolutions and furrows. [ 1 ]
In the complete absence of convolutions, agyria is defined, and the presence of several wide flat convolutions is called pachygyria. These defects, like some other reduction deformations of the brain, have the code Q04.3 in ICD-10.
Epidemiology
According to statistics on rare diseases, there are 1-1.2 cases of lissencephaly per 100 thousand newborns. [ 2 ], [ 3 ]
According to some data, up to 25-30% of cases of classical lissencephaly are observed in children with Miller-Dieker syndrome; point mutations and deletions of the LIS1 and DCX genes are detected in almost 85% of patients. [ 4 ]
Genetic studies of 17 genes associated with lissencephaly showed that LIS1 mutation or deletion accounts for 40% of patients, and 23% are associated with DCX mutation, followed by TUBA1A (5%) and DYNC1H1 (3%).[ 5 ]
Causes lissencephaly
All known causes of the formation of the cerebral cortex (cortex cerebri) almost or completely without convolutions and furrows, which increase the "working area" of the human brain and ensure the "productivity" of the central nervous system, are associated with disturbances in its perinatal development. That is, lissencephaly develops in the fetus. [ 6 ]
The failure to form layers of the cerebral cortex of the fetus in lissencephaly is the result of abnormal migration of the neurons that form it or premature cessation of this process.
This process, which is essential for cerebrocortical histogenesis, occurs in several stages from the 7th to the 18th week of pregnancy. And, given its increased sensitivity to genetic mutations, as well as various negative physical, chemical and biological influences, any deviation from the norm can lead to incorrect localization of neurons with the possible formation of a thickened layer of gray matter of the cortex without a characteristic structure. [ 7 ]
In some cases, lissencephaly in children is associated with Miller-Dieker, Walker-Warburg, or Norman-Roberts syndromes.
Read also – Developmental defects of the brain
Risk factors
In addition to mutations in some genes, risk factors for the birth of a child with such a severe defect include oxygen starvation (hypoxia) of the fetus; insufficient blood supply to the brain (hypoperfusion); acute cerebrovascular accident in the form of perinatal stroke; placental pathologies; viral infections of the pregnant woman (including TORCH); [ 8 ] problems with general metabolism and thyroid function; smoking, alcohol, psychotropic and narcotic substances; use of a number of medications; increased radiation levels. [ 9 ]
Pathogenesis
Not all cases of lissencephaly have a pathogenesis caused by chromosomal abnormalities and gene mutations. But some genes are known that encode proteins that play an important role in the correct movement of neuroblasts and neurons along the radial glia cells - to form the cerebral cortex. And mutations of these genes lead to this pathology. [ 10 ]
In particular, these are sporadic mutations (without heredity) of the LIS1 gene on chromosome 17, which regulates the cytoplasmic motor protein of microtubules dynein, as well as the DCX gene on the X chromosome, which codes for the protein doublecortin (lissencephalin-X). [ 11 ]. In the first case, specialists define classical lissencephaly (type I), in the second – X-linked. [ 12 ]
When the FLN1 gene, which encodes the phosphoprotein filamin 1, is deleted, the process of directed neuronal migration may not begin at all, leading to a complete absence of convolutions (agyria). [ 13 ]
Mutations have been identified in the CDK5 gene, which encodes a kinase enzyme – a catalyst for intracellular metabolism, regulating the cell cycle in CNS neurons and ensuring their normal migration during the prenatal formation of brain structures.
Abnormal changes in the RELN gene on chromosome 7 that cause cortical gyral defects in Norman-Roberts syndrome result in a deficiency of the extracellular glycoprotein reelin, which is required to regulate the migration and positioning of neural stem cells during cortex cerebri development.[ 14 ], [ 15 ], [ 16 ]
The ARX gene encodes the non-aristalens homeobox protein, a transcription factor that plays an important role in the forebrain and other tissues.[ 17 ] Children with the ARX mutation have other symptoms, such as missing parts of the brain (agenesis of the corpus callosum), abnormal genitalia, and severe epilepsy.[ 18 ],[ 19 ]
Several genes have been linked to lissencephaly. These genes include VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A, and CDK5.[ 20 ]
Cytomegalovirus (CMV) has been associated with the development of lissencephaly due to decreased blood supply to the fetal brain. The severity of CMV infection depends on the gestational age. Early infection is more likely to cause lissencephaly because neuronal migration occurs early in pregnancy.[ 21 ]
In addition, the mechanism of occurrence of this anomaly includes incomplete or later cessation of migration of neurons from the periventricular generative zone to the cerebral cortex. And in such cases, either incomplete lissencephaly or pachygyria develops, in which several wide grooves and convolutions are formed (but most of them are absent).
Symptoms lissencephaly
The first signs of this pathology (in the absence of the previously mentioned syndromes) may not appear immediately after birth, but after one and a half to two months. And most often, the following clinical symptoms of lissencephaly are observed:
- muscular hypotonia, often combined with spastic paralysis;
- convulsions and generalized tonic-clonic seizures (in the form of opisthotonus);
- severe mental retardation and growth retardation;
- impairment of neurological and motor functions.
Problems with swallowing cause difficulties in feeding the baby. [ 22 ]
A high degree of neuromotor impairment often manifests itself as tetraplegia – paralysis of all limbs. Deformation of the hands, fingers or toes is possible.
In Norman-Roberts syndrome with lissencephaly type I, craniofacial anomalies are observed: severe microcephaly, low forehead slope and protruding broad bridge of the nose, wide-set eyes (hyperterlorism), underdevelopment of the jaws (micrognathia). [ 23 ]
Miller-Dieker syndrome may also be characterized by an abnormally small head size with a wide, high forehead and short nose, depressions in the temples (bitemporal depressions), and low-set, deformed ears.
Severe lissencephaly syndrome is characterized by microcephaly, a decrease in the size of the eyeballs (microphthalmia), combined with retinal dysplasia, obstructive hydrocephalus, and absent or hypoplastic corpus callosum.
Complications and consequences
Among the complications of this anomaly, specialists name a violation of the swallowing function (dysphagia) and gastroesophageal reflux; refractory (uncontrolled) epilepsy; frequent upper respiratory tract infections; pneumonia (including chronic aspiration).
Infants with lissencephaly may have congenital organic cardiac problems in the form of an atrial septal defect or a complex heart defect with cyanosis (tetralogy of Fallot). [ 24 ]
The consequences of postnatal growth retardation in most cases result in death within 24 months after birth.
Diagnostics lissencephaly
Diagnosis begins with a physical examination of the child, a study of the parents' medical history and the history of pregnancy and childbirth.
During gestation, cell-free fetal DNA testing, amniocentesis, or chorionic villus sampling may be required. [ 25 ] For more information, see – Prenatal Diagnosis of Congenital Diseases
Instrumental diagnostics are used to visualize brain structures and assess their functions:
- computed tomography of the brain;
- magnetic resonance imaging (MRI) of the brain;
- electroencephalogram (EEG). [ 26 ]
During pregnancy, lissencephaly on ultrasound of the fetus after 20-21 weeks may be suspected in the absence of the parieto-occipital and calcarine grooves and an anomaly of the Sylvian fissure of the brain.
Differential diagnosis
Differential diagnostics with other syndromes of congenital cerebral defects is carried out.
There are more than 20 types of lissencephaly, most of which fall into 2 main categories: classic lissencephaly (type 1) and cobblestone lissencephaly (type 2). Each category has similar clinical manifestations but different genetic mutations.[ 27 ]
Brain examination in type I lissencephaly shows a cerebral cortex with four layers instead of the six seen in normal patients, whereas in type 2 lissencephaly the cerebral cortex is disorganized and appears lumpy or nodular due to complete displacement of the cerebral cortex by clusters of cortical neurons separated by gliomesenchymal tissue. Patients also had muscle and eye abnormalities.
- Classic lissencephaly (type 1):
- LIS1: Isolated lissencephaly and Miller-Dieker syndrome (lissencephaly associated with facial dysmorphism). [ 28 ]
- LISX1: DCX gene mutation. Compared to lissencephaly caused by LIS1 mutations, DCX shows a six-layer cortex instead of four.
- Isolated lissencephaly without other known genetic defects
- Cobblestone lissencephaly (type 2):
- Walker-Warburg syndrome
- Fukuyama Syndrome
- Disease of muscles, eyes and brain
- Other types cannot be placed into one of the two above groups:
- LIS2: Norman-Roberts syndrome, similar to lissencephaly type I or Miller-Dieker syndrome, but without the deletion of chromosome 17.
- LIS3
- LISX2
Microlissencephaly: This is a combination of the absence of normal cortical folding and an abnormally small head. Children with regular lissencephaly have normal-sized heads at birth. Children with a small head size at birth are usually diagnosed with microlissencephaly.
It is also important to distinguish between lissencephaly and polymicrogyria, which are different brain developmental defects.
Who to contact?
Treatment lissencephaly
Lissencephaly is an incurable organic defect, so only supportive and symptomatic treatment is possible. [ 29 ]
First of all, this is the use of anticonvulsants and antiepileptic drugs, as well as the installation of a gastrostomy tube in the stomach (if the child is unable to swallow independently). Massage is useful.
In cases of severe hydrocephalus, cerebrospinal fluid is removed.
Prevention
Experts recommend that future parents seek genetic counseling, and that pregnant women register with obstetricians and gynecologists in a timely manner and undergo all scheduled examinations.
Forecast
For children with lissencephaly, the prognosis depends on its degree, but most often the child's mental development does not exceed the level of four to five months. And all children with this diagnosis suffer from severe psychomotor disorders and difficult-to-treat epilepsy. [ 30 ]
According to NINDS (the American National Institute of Neurological Disorders and Stroke), the maximum life expectancy for people with lissencephaly is about 10 years.