
All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Rabies in children
Medical expert of the article

Last reviewed: 04.07.2025
Rabies, or hydrophobia, is an acute viral disease transmitted through the bite of an infected animal, with damage to the nervous system and the development of severe encephalitis with a fatal outcome.
Epidemiology
A public health scourge since ancient times, the rabies virus currently causes approximately 59,000 human deaths each year, almost all of which are transmitted by dog bites. This has a significant economic impact on developing countries, particularly in Africa and Asia, which can bear the least such losses. However, despite its nearly 100% fatality rate, canine rabies is an entirely preventable disease, and historical examples of canine rabies eradication in the developed world attest to this. [ 1 ]
Causes rabies
The causative agent is the rabies virus (RV), a negative-strand RNA virus of the rhabdovirus family, approximately 60 nm × 180 nm in size.
It consists of an inner protein core, or nucleocapsid, containing nucleic acid, and an outer membrane, a lipid-containing bilayer covered with transmembrane glycoprotein spikes. It has a relatively simple modular genome structure and encodes five structural proteins:
- RNA-dependent RNA polymerase (L),
- nucleoprotein (N),
- phosphorylated protein (P),
- matrix protein (M) and
- outer surface glycoprotein (G).
The N, P, and L proteins together with the genomic RNA form the ribonucleoprotein complex. G is the only RV antigen capable of inducing the production of RV neutralizing antibodies, which are the major immune effectors against lethal RV infection. On the other hand, the ribonucleoprotein complex has been shown to be the major RV antigen capable of inducing CD4+ T cells, which can enhance the production of RV neutralizing antibodies through intrastructural antigen recognition.[ 2 ] The ribonucleoprotein complex may play an important role in the establishment of immunological memory and long-term immunity.[ 3 ]
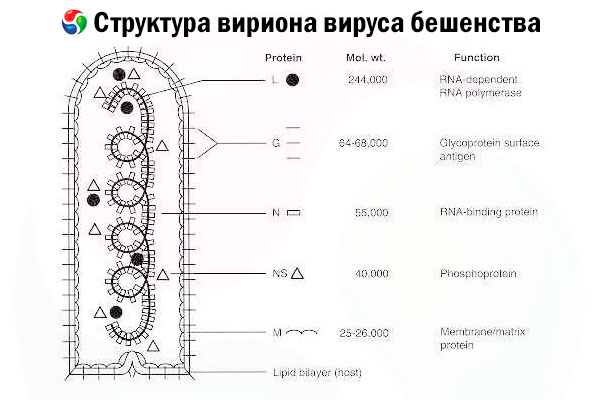
 [
[ Classification and antigen types
The genus Lyssavirus includes rabies virus and antigenically and genetically related rabies viruses: Lagos, Mokola, and Duvenhage bat viruses, as well as two putative subtypes of European bat lyssaviruses. Cross-protection studies indicate that animals immunized with traditional rabies vaccines may not be fully protected when challenged with other lyssaviruses.
Rabies viruses can be classified as fixed (adapted by passage in animals or cell culture) or street (wild type). The use of monoclonal antibodies and genetic sequencing to differentiate street rabies viruses has helped in identifying viral variants originating from major host reservoirs worldwide and in suggesting likely sources of human exposure when a history of a definitive animal bite was otherwise absent in a patient's case.[ 8 ]
Pathogenesis
The main reservoir and source of infection among wild animals are wolves, foxes, jackals, bats, and among domestic animals - dogs and cats, rarely - horses, cattle, pigs, rats, etc. Transmission of infection from person to person, although possible, is extremely rare. This is a typical zoonotic infection. People become infected with rabies mainly from dogs.
After a human is bitten by a sick animal, the virus multiplies in the muscle tissue at the site of the bite, and then, having reached the ends of the sensory peripheral nerves, spreads centripetally, reaching the motor neurons. The time it takes for the virus to move and the brain to be affected depends on the site of the bite. In case of severe bites of the head and face, the virus can reach the central nervous system in 15-20 days, and in case of minor damage to the skin of the trunk and limbs and, consequently, a small dose of the pathogen, the process of moving the virus to the central nervous system can be delayed for several months or even up to 1-1.5 years. Having reached the central nervous system, the virus is fixed in the tissues of the brain and spinal cord, mainly in the neurons of the medulla oblongata, the Ammon's horn, and the base of the brain. In the spinal cord, the posterior horns are most affected. From the central nervous system, the virus centrifugally along the nerve trunks reaches the salivary glands, where it multiplies and is excreted with saliva.
Concepts in the pathogenesis of rabies
RV has a broad host range and can infect almost all mammals. Although several routes of RV transmission have been reported, natural infection most commonly occurs via a bite. In addition to bites, consumption of RV-infected carcasses can promote rabies virus infection in Arctic foxes, and contact of RV with mucous membranes has been found to be another possible route of transmission.[ 9 ] In some unusual circumstances, such as accidental release of RV as an aerosol in a laboratory or RV as an aerosol in caves inhabited by large numbers of bats,[ 10 ] aerosol transmission may occur.
It is not yet clear whether street RV and mouse-adapted or tissue culture-adapted RV strains replicate at the site of inoculation before they enter the CNS. While experimental intramuscular infection of juvenile hamsters or raccoons with street RV revealed RV replication in striated muscle cells before the virus invaded motor neuron axons across neuromuscular junctions,[ 11 ],[ 12 ] intramuscular infection of mice with mouse-adapted CVS-24 RV showed that RV migrates directly to the CNS without prior replication at the site of inoculation.[ 13 ] Once in the terminals of unmyelinated axons, RV is retrogradely transported to the cell body.
Recent findings suggest that axonal vesicle transport may represent a key strategy for long-distance virion movement in axons.[ 14 ] It has been estimated that RV migrates within axons at a rate of 3 mm/h.[ 15 ] The infection then spreads through a chain of neurons connected by synaptic junctions. However, the exact mechanism that promotes transsynaptic spread is still unknown. After infecting the brain, the virus spreads centrifugally to the peripheral and autonomic nervous system in many peripheral organs.[ 16 ] In the last stage of the infection cycle, RV migrates to the salivary glands; after replication in mucogenic acinar cells, it is released into saliva and is ready for transmission to the next host.[ 17 ]
With regard to rabies virus-induced pathology, apoptotic cell death has been proposed as a potential pathogenic mechanism in experimental rabies models of mice infected with a fixed strain of RV.[ 18 ] A pathogenic mechanism that may contribute to the profound CNS dysfunction characteristic of rabies may be impaired neuronal function. Gene expression has been shown to be markedly reduced in RV-infected neurons, resulting in a general suppression of protein synthesis,[ 19 ] and several studies have shown impaired neurotransmission following RV infection. Jiang demonstrated that binding of an acetylcholine receptor antagonist to infected rat brain homogenates was reduced compared with controls.[ 20 ] Impaired release and binding of serotonin, a neurotransmitter involved in the control of the sleep cycle, pain perception, and behavior, were also observed in RV-infected rat brain. [ 21 ], [ 22 ] In addition to affecting neurotransmission, right ventricular infection may also affect ion channels. Infected mouse neuroblastoma cells exhibit decreased functional expression of voltage-gated sodium channels, which may prevent action potentials and ultimately lead to functional impairment. [ 23 ]
In addition to the absence of serious pathological lesions in the CNS, most cases of human rabies do not elicit an immune response 7 to 10 days after the onset of clinical signs. These profound differences between the pathogenesis of rabies and that of most other viral or bacterial CNS infections are further supported by the fact that immunosuppression is either ineffective or detrimental to the outcome of rabies.[ 24 ] The low level of immune response often observed in rabies victims is puzzling because it cannot be explained by the poor immunogenicity of RV antigens. In fact, RV G and nucleocapsid protein are potent B- and T-cell antigens when administered parenterally. [ 25 ] A possible explanation for the low degree of immune response against RV in humans or animals with rabies may be that RV infection of the CNS causes immunosuppression, [ 26 ] and it has been proposed that RV uses a subversive strategy including preventing apoptosis and destroying invading T cells. [ 27 ]
Attenuated RV strains that have been adapted to non-neuronal cells differ significantly from pathogenic street RV strains in their neuroinvasiveness, which refers to their ability to invade the CNS from peripheral sites. In this regard, tissue culture-adapted RV strains either lack or have only limited ability to invade the CNS from peripheral sites, whereas street RV strains or mouse-adapted RV strains such as CVS-24 are highly invasive.[ 28 ] Key factors involved in RV neuroinvasion include viral uptake, axonal transport, trans-synaptic spread, and viral replication rate.
Until recently, our knowledge of RV pathogenesis was limited and was based primarily on descriptive studies of street RV strains or experimental infections with attenuated strains adapted in the laboratory. The advent of reverse genetics technology has allowed us to identify the viral elements that determine the pathogenic phenotype of RV and to better understand the mechanisms involved in rabies pathogenesis.
Identification of viral elements controlling rabies virus acquisition, dissemination and replication
- Viral elements involved in virus capture
RV infection begins with attachment of the virus to a putative cellular receptor. Although several membrane surface molecules have been proposed as RV receptors, including the nicotinic acetylcholine receptor,[ 29 ] the neural cell adhesion molecule[ 30 ] and the low-affinity neurotrophin receptor p75 NTR,[ 31 ] it is still unclear whether these molecules actually play a role in the rabies virus life cycle. In this context, it has recently been shown that the RV G–p75 NTR interaction is not required for RV infection of primary neurons.[ 32 ] Following receptor binding, RV is internalized via adsorptive or receptor-mediated endocytosis. [ 33 ] The low pH environment within the endosomal compartment then induces conformational changes in RV G that trigger fusion of the viral membrane with the endosomal membrane, thereby releasing the RNP into the cytoplasm. [ 34 ] For viruses, RV G plays a critical role in viral uptake, most likely through interactions with putative cellular receptors that facilitate rapid uptake. In this regard, it has been demonstrated that the pathogenicity of tissue culture-adapted RV strains (e.g., ERA, HEP, and CVS-11) correlates with the presence of a determinant located in antigenic site III of the G protein. [ 35 ] An Arg → Gln mutation at position 333 in this antigenic site of the ERA G protein resulted in a seven-fold delay in the internalization of the Gln333 RV variant compared to the wild-type variant. The Asn194→Lys194 mutation in RV G, which explains the re-emergence of the pathogenic phenotype, was associated with a significant decrease in internalization time.[ 36 ] Furthermore, experiments with chimeric RVs showed that the time required for internalization of RV virions was significantly increased and pathogenicity was strongly reduced following replacement of the G gene of the highly pathogenic SB RV strain, which was derived from a cDNA clone of the silver-derived bat-associated strain RV-18,[ 37 ] with that of the highly attenuated SN strain, which was isolated from a cDNA clone of the SAD B19 RV vaccine strain.[ 38 ] Together, these data support the notion that the kinetics of virus uptake, which is a function of RV G, is a major determinant of RV pathogenicity.
- Viral elements involved in the spread and transmission of viruses
A unique property of rabies virus is its ability to spread from cell to cell. The observation that the Gln333 ERA variant loses pH-dependent cell-cell fusion activity in vitro [ 39 ] and displays a greatly reduced ability to spread from cell to cell [ 40 ] suggests that RV G also plays a key role in cell-to-cell spread and hence virus transmission, likely through its fusiogenic activity. This possibility is further supported by the finding that the spread rate of the pathogenic RV revertant SPBNGAK is almost twice as high as that determined for the non-pathogenic SPBNGA variant. Interestingly, the Asn 194 → Lys 194 mutation in G SPBNGAK caused a shift in the pH threshold for membrane fusion to a higher pH, supporting the hypothesis that a higher pH threshold for membrane fusion is associated with increased virus spread. [ 41 ]
Studies of transneuronal indicators of RV infection in rats [ 42 ] and rhesus monkeys [ 43 ] have shown that rabies virus migrates exclusively in a retrograde direction in axons. Although several RV proteins are involved in neuronal transport mechanisms, RV G appears to play a predominant role in transneuronal spread of RV infection. For example, while peripheral infection with equine infectious anemia virus (EIAV) pseudotyped with RV G results in viral transfer to the spinal cord, the same EIAV pseudotyped with vesicular stomatitis virus G failed to enter the nervous system. [ 44 ] Furthermore, viral spread of the ERA G Arg 333 → Gln 333 mutant in the CNS was found to be strongly reduced compared to the wild-type mutant, further suggesting a function of intact RV G in trans-synaptic spread. However, the most compelling evidence for an important role of RV G in trans-synaptic transport comes from intracranial infection of mice with a recombinant G-deficient RV virus, which showed that the infection remained restricted to neurons at the site of inoculation without any evidence of spread to secondary neurons.[ 45 ] However, it is likely that in addition to RV G, RV M also plays a role in virus spread and hence in trans-synaptic transport. In this regard, it was shown that the spread of the chimeric SN-BMBG RV variant, which contains both M and G from the highly pathogenic SB, was significantly higher than the spread of the chimeric SN-BG or SN-BM variant, which contain the G and M from SB, respectively, suggesting that optimal interaction of M with G may play an important role in cell-to-cell virus spread. [ 46 ] Since RV M supports virus budding, [ 47 ] it is likely that the more efficient spread of the RV SN-BMBG chimeric variant is due to optimal virus budding at the postsynaptic membrane.
Recent studies have shown that the interaction between RV P and the dynein light chain links the RV RNP to the host cell transport system, thereby facilitating retrograde axonal transport of the virus.[ 48 ],[ 49 ] However, peripheral infection of adult mice showed that deletion of the LC8 binding domain of RV P does not prevent virus entry into the CNS, suggesting that the RV protein is not directly involved in retrograde axonal spread of RV.[ 50 ]
- Viral elements that control viral replication
Unlike many other viruses, such as influenza virus, RV pathogenicity is inversely proportional to the rate of viral RNA synthesis and production of infectious viral particles. Comparison of viral mRNA and genomic RNA levels produced by different chimeric viruses suggests that viral RNA transcription and replication are regulated by multiple factors, including RV M, which has been identified as a trans-acting factor that mediates the switch from initial high levels of mRNA synthesis to genomic RNA synthesis.[ 51 ] Furthermore, M from all rhabdoviruses is able to shut off viral gene expression by binding to the RNP, resulting in the formation of a highly condensed backbone-like structure that is unable to support RNA synthesis.
To identify other viral elements that control pathogenicity by regulating viral replication, the 5' terminal sequences of the highly pathogenic SB strain were replaced stepwise with sequences from the highly attenuated SN vaccine strain, resulting in recombinant viruses SB2 (terminal sequence [TS] + L), SB3 (TS + L + pseudogene [Ψ]), SB4 (TS + L + Ψ + G), and SB5 (TS + L + Ψ + G + M). Intramuscular infection with the parental SB and SN viruses and the chimeric RVs SB2, SB3, SB4, and SB5 elicited the highest mortality rates in SB-infected mice and no morbidity or mortality in SN-infected mice. Replacement of TS, L, and SB with the corresponding elements from SN resulted in a modest reduction in morbidity and mortality, and an additional G or G plus M exchange strongly reduced or completely abolished viral pathogenicity.
Phenotypic characterization of these wild-type and chimeric RVs in tissue culture revealed that the pathogenicity of a given RV is inversely correlated with its ability to replicate in neuronal cells. Although SB replicated at levels nearly 1000-fold lower than SN, and replacement of TS, L, and in SB by SN levels had little effect on viral growth kinetics, additional replacement of the G or G plus M of SB by the corresponding SN genes resulted in a 1-log increase in virus production, suggesting that viral RNA replication kinetics as well as viral particle production are largely controlled by the RV G protein. This conclusion is supported by data obtained with RV G variants that differ by one amino acid in their G proteins. The pathogenic rabies virus variant SPBNGAK 194 produced a virus titer in NA cells that was 1 log lower than that produced by the nonpathogenic variant SPBNGAN 194, and real-time PCR analysis showed that the rates of viral RNA transcription and replication in SPBNGAK-infected NA cells were 5- and 10-fold higher than in SPBNGAK-infected NA cells.[ 52 ] Further evidence for an inverse correlation between pathogenicity and the rate of viral RNA synthesis and viral particle production was provided by mice infected with chimeric recombinant viruses in which the G and M genes of the attenuated SN strain were replaced by those of the highly pathogenic SB strain. These experiments revealed a significant increase in the pathogenicity of the parental SN strain carrying RV G over the pathogenic SB strain. Pathogenicity was further increased when both G and M from SB were introduced into SN.
Substitution of G or M or both in SN with the corresponding genes from SB was associated with a significant decrease in the rate of viral particle production as well as the rate of viral RNA synthesis. These data indicate that both G and M play important roles in RV pathogenesis by regulating viral replication. The finding that substitution of G or G plus M in SN with G or G plus M of SB results in a moderate to strong decrease in viral RNA transcription and replication, respectively, while substitution of M alone in SN with M of SB results in a strong increase in viral RNA transcription and replication, indicates that RV G also has an important regulatory function in viral RNA transcription/replication either alone or through interaction with the M protein. The mechanism by which the RV G gene controls viral RNA synthesis is unknown. Certain nucleotide sequences within the RV G genes, such as those including the codons for Arg333 and Lys 194, have been identified as targets for cellular miRNAs. It has been shown that target recognition by cellular miRNAs can result in positive or negative regulation of viral replication. [ 53 ] Arg 333 → Glu 333 or Lys 194 → Ser 194 substitutions within the RV G gene sequence result in the abolition of miRNA target sequences, which in turn is associated with a significant increase in the rate of viral RNA synthesis [Faber M, Thomas Jefferson University, PA, USA, Unpublished Data], suggesting that host cellular miRNAs also play an important role in the regulation of RV replication, as has been shown for other RNA viruses including vesicular stomatitis virus and HCV. [ 54 ], [ 55 ]
Regulation of viral replication appears to be one of the important mechanisms involved in RV pathogenesis. To evade the immune response and preserve the integrity of the neuronal network, pathogenic RV strains, but not attenuated strains, can regulate their growth rate. A lower rate of replication likely benefits pathogenic RV strains by preserving the neuronal structure that these viruses use to reach the CNS. Another explanation for the lower replication rate of pathogenic RV is that, in order to evade early detection by the host immune system, the virus maintains minimal levels of expression of its antigens.
Relationship between RV G expression, apoptosis and pathogenicity
It is well known that street rabies virus strains that are significantly more pathogenic than tissue culture-adapted strains express very limited levels of G and do not induce apoptosis until late in the infectious cycle, suggesting that the pathogenicity of a particular virus strain is inversely correlated with RV G expression and the ability to induce apoptosis.[ 56 ] Direct evidence for a correlation between the level of G expression and the extent of apoptosis was obtained with the recombinant RV SPBNGA-GA, which carried two identical G genes and overexpressed RV G.[ 57 ] Morphological studies of neuronal cultures infected with this recombinant RV showed that cell death was significantly increased in parallel with RV G overexpression and that apoptosis is the major mechanism involved in RV G-mediated death. In particular, the decrease in F-actin staining after SPBNGA-GA infection is consistent with apoptosis-induced depolymerization of actin filaments. Furthermore, the number of TUNEL-positive nuclei in SPBNGA-GA-infected neurons was significantly increased compared to that in uninfected and SPBNGA-infected neurons. However, the mechanism by which RV G gene mediates the apoptotic signaling process remains largely unknown. It has been suggested that RV G expression above a certain threshold severely disrupts the cell membrane. It is highly likely that apoptotic cells are not cleared quickly in the CNS and therefore undergo secondary necrosis. [ 58 ] On the other hand, RV infection and in particular RV G protein overexpression can lead to pyroptosis, a cell death pathway similar to apoptosis that, unlike apoptosis, involves the activation of caspase 1 and thereby leads to necrosis. [ 59 ] The degree of necrosis or pyroptosis induced by RV infection likely plays a critical role in the induction of antiviral immunity. While apoptotic cells maintain their membrane integrity and do not stimulate the innate immune response, necrotic cells become permeabilized and secrete endogenous adjuvants that can trigger a robust innate immune response. [ 60 ]
Since the level of apoptosis/necrosis correlates with RV immunogenicity, it has been suggested that the immunostimulatory effect of apoptotic/necrotic cells most likely contributes to the generation of a protective immune response. Therefore, regulation of RV G expression is very likely an important factor in rabies pathogenesis, as it provides a means for the survival and dissemination of pathogenic RV variants in the nervous system without causing overt neuronal damage and eliciting a protective immune response that would prevent infection.
RV G expression may be regulated at the level of RNA synthesis, the post-translational level, or both. The levels of RV G expressed by different RV chimeric variants have been shown to be reflected by the rate of viral RNA synthesis, suggesting that differential regulation of RV G expression by these variants results from variations in the rate of viral mRNA transcription. As with viral RNA transcription rates, the amount of RV G expressed by these variants inversely correlates with viral pathogenicity. On the other hand, infection of primary neuronal cultures with the less pathogenic RV variant CVS-B2c resulted in fourfold higher levels of G protein than infection with the highly pathogenic variant CVS-N2c, despite the synthesis of comparable levels of G mRNA in both infections. Pulse-chase experiments showed that the higher G protein levels in CVS-B2c-infected neurons were largely the result of a lower rate of degradation of the CVS-B2c G protein compared to the CVS-N2c G protein. However, the mechanism that leads to the more rapid proteolytic degradation of the CVS-N2c G protein remains to be elucidated.
Symptoms rabies
The incubation period for rabies is on average 30-90 days. In case of massive infection through large wounds of the head and face, it can be shortened to 12 days. In rare cases, the incubation period can last 1 year or more.
There is a strictly sequential change of three periods of the disease: prodromal, excitation, paralysis.
The prodromal period begins with the appearance of aching or pulling pain at the site of the bite, as well as pain along the nerves. In the area of the scar, there may be a burning sensation, itching, sometimes redness and swelling. The patient experiences general malaise, headache, nausea. Vomiting, an increase in body temperature to 37.5-38 ° C and symptoms of a progressive mental disorder are noted: increased reflex excitability, an inexplicable feeling of anxiety, fear, melancholy. Often the patient is depressed, inhibited, withdrawn, refuses to eat, sleeps poorly, complains of gloomy thoughts, frightening dreams. The prodromal period lasts 2-3 days, sometimes extends to 7 days. At the end of this period, there may be attacks of anxiety with short-term breathing difficulties, a feeling of tightness in the chest, accompanied by tachycardia and increased respiratory rate.
The period of excitement is marked by the appearance of hydrophobia: when trying to drink, and then at the sight of water or a reminder of it, the patient experiences a convulsive spasm of the pharynx and larynx, during which he throws away the mug of water with a scream, throws forward trembling hands, throws back his head and body. The neck is stretched out, a painful grimace distorts the face, which becomes bluish due to a spasm of the respiratory muscles. The eyes bulge, express fear, beg for help, the pupils are dilated, inhalation is difficult. At the height of the attack, cardiac and respiratory arrest is possible. The attack lasts for several seconds, after which the patient's condition seems to improve. Subsequently, attacks of spasms of the muscles of the larynx and pharynx can occur even from the movement of air (aerophobia), bright light (photophobia) or a loud word (acousticophobia). The attacks are accompanied by psychomotor agitation, during which the patient behaves like a "madman". Consciousness is clouded during the attack, but clears up in the interictal period. During the period of agitation, due to the increased tone of the sympathetic nervous system, patients experience a sharp increase in salivation (sialorrhea) with the inability to swallow saliva due to spasm of the pharyngeal muscles. The patient sprays saliva. Some patients may develop signs of meningism and even opisthotonus, and convulsions are common. In this case, the cerebrospinal fluid may not change, but in some patients, the protein concentration may increase and the number of cells may increase due to lymphocytes.
Without adequate treatment, signs of dehydration increase, facial features become sharper, and body weight decreases. Body temperature rises to high values. Convulsions are possible. The duration of the excitation stage is about 2-3 days, rarely 4-5 days. A fatal outcome usually occurs during one of the attacks. Rarely, the patient survives to the third stage of the disease.
During the period of paralysis, the patient calms down. Attacks of hydrophobia cease, the patient can drink and swallow food, consciousness is clear. However, despite the apparent well-being, lethargy, apathy, depression increase, paralysis of the limbs, pelvic disorders, paralysis of the cranial nerves soon appear. Body temperature rises to 42-43 °C, arterial pressure drops, and by the end of the first day death occurs from paralysis of the cardiovascular and respiratory centers.
Neutrophilic leukocytosis, increased hemoglobin, erythrocytes, and hematocrit are observed in the peripheral blood.
What's bothering you?
Forms
Clinically, typical and atypical forms are distinguished. Atypical forms include all cases without arousal and hydrophobia. Atypical forms include bulbar, cerebellar, meningoencephalitic, etc.
Diagnostics rabies
Detection of rabies antigen, antibodies, viral RNA, or virus isolation allows diagnosis of rabies. Because any individual test may be negative in a patient with rabies, serial serum samples for rabies antibody detection, saliva samples for viral culture, and skin biopsy for direct immunofluorescence testing for viral antigen are sometimes necessary, especially when rabies is highly suspected.
One of the most rapid methods for diagnosing antemortem rabies in humans is to perform a direct immunofluorescence test on a skin biopsy of the nape of the neck to detect rabies antigen. The direct immunofluorescence test is the most sensitive and specific method for detecting rabies antigen in skin and other fresh tissues (e.g., brain biopsy), although results may occasionally be negative early in the disease. If fresh tissue is unavailable, enzymatic digestion of fixed tissues may increase the reactivity of the immunofluorescence test; however, sensitivity may be unacceptably low.
The diagnosis can also be established if the virus is isolated from saliva after inoculation of neuroblastoma cells or laboratory rodents; this is usually most effective during the first 2–3 weeks of illness. Detection of rabies virus-neutralizing antibodies, usually performed by the rapid fluorescent focus inhibition test (RFFIT), in the serum of unvaccinated individuals is also diagnostic. The presence of antibodies in the cerebrospinal fluid confirms the diagnosis, but they may appear 2–3 days later than serum antibodies and may therefore be less useful in the early stages of the disease. While the serologic response following vaccination is generally indistinguishable from the serologic response induced by the disease, vaccination does not usually produce antibodies to the cerebrospinal fluid.
Only seven cases of rabies "recovery" in the last 25 years have been well documented. Although rabies virus was not isolated from any of the patients, high titers of rabies-neutralizing antibodies in serum samples and the presence of neutralizing antibodies in the cerebrospinal fluid strongly supported the diagnosis.
What do need to examine?
What tests are needed?
Differential diagnosis
The diagnosis of human rabies is usually made on the basis of epidemiologic and clinical data and confirmed in the laboratory. The diagnosis is straightforward if there is a history of animal bites and the full spectrum of symptoms and signs has occurred. Otherwise, a careful but rapid evaluation of the epidemiologic and clinical features of less typical cases is necessary before performing specific laboratory tests. Any patient with neurologic signs or symptoms or unexplained encephalitis should be questioned about the possibility of exposure to animals in rabies-endemic areas within or outside the country of residence. The failure to suspect rabies in several recent human deaths in the United States may have been due to a lack of careful history of exposure.
At the onset of the disease, rabies can mimic many infectious and non-infectious diseases. Many other encephalitides, such as those caused by herpesviruses and arboviruses, resemble rabies. Other infectious diseases can also mimic rabies, such as tetanus, cerebral malaria, rickettsiosis, and typhoid fever. Paralytic infectious diseases that can be confused with rabies include poliomyelitis, botulism, and herpes simian B encephalitis.
Non-infectious diseases that may be confused with rabies include a number of neurological syndromes, especially acute inflammatory polyneuropathy (Guillain-Barré syndrome), as well as allergic post-vaccination encephalomyelitis secondary to rabies vaccination of the nervous tissue, poisoning or drug intoxication, alcohol withdrawal, acute porphyria, and rabies hysteria. Guillain-Barré syndrome may be mistaken for paralytic rabies, and vice versa.
Who to contact?
Treatment rabies
Treatment for rabies has not been developed. Administration of large doses of specific anti-rabies immunoglobulin and leukocyte interferon is ineffective. Symptomatic treatment is administered to alleviate the patient's suffering. For this purpose, the patient is placed in a separate ward or box, a protective regime is created that limits the influence of the external environment (reduced noise, bright light, air flow). To reduce the excitability of the central nervous system, sleeping pills, anticonvulsants, and painkillers are prescribed. The water balance is normalized.
In the paralytic stage, drugs are prescribed that stimulate the activity of the cardiovascular and respiratory systems. It is recommended to use hyperbaric oxygenation, cerebral hypothermia, controlled mechanical breathing with complete curarization of the patient. However, all treatment methods are practically ineffective. In the best case, it is possible to prolong the patient's life for several months. An unfavorable outcome is predetermined by the severity of the damage to the brain stem with the destruction of vital centers.
Prevention
The development of the first rabies vaccine by Pasteur in 1885 ushered in an era of much more effective rabies control. Today, despite the nearly 100% mortality rate in humans from rabies, the disease is completely preventable through pre- and/or post-exposure vaccination. While Pasteur and his colleagues initiated the vaccination of private dogs in Paris, the first mass vaccination of dogs was carried out in the early 1920s in Japan, marking the first major national rabies control program. Oral vaccination of wild animals, first developed in the 1970s, has since been repeatedly shown to effectively control the disease in major terrestrial hosts such as foxes, raccoons, and skunks.[ 68 ] Sustained rabies vaccination of reservoir animal populations at 70% or higher coverage rates will eventually eliminate RABV from reservoir species and prevent the spread of the virus to incidental hosts. [ 69 ]
Phylogenetic data indicate that lyssaviruses infected bats long before they infected terrestrial mammals, and most lyssaviruses, including RABV, still circulate in various bat species worldwide.[ 70 ] However, effective methods to prevent transmission of RABV among bats remain elusive, precluding the possibility of complete rabies eradication at this time. However, even after exposure to RABV through the bite of a rabies-infected mammal, safe and effective post-exposure prophylaxis (PEP, including wound cleaning, rabies immune globulin, and rabies vaccination) can protect humans from rabies infection if treatment is administered promptly and according to World Health Organization (WHO) recommendations.
These two methods of preventing human deaths—one based on vaccinating exposed people and the other based on vaccinating enough dogs to break the cycle of transmission at the source—are the building blocks of a "one health" approach to canine rabies prevention and control. These two different means of preventing human deaths were considered as separate alternatives: Strategy A, based on providing PEP to people, and Strategy B, based on vaccinating dogs; or as components of a combined Strategy A + B in an analysis of the likely costs of the alternative strategies.[ 71 ]
Countries such as Thailand have had enormous success in preventing human deaths through the use of PEP, but have also found increasing demand and associated costs associated with the use of PEP alone. [ 72 ] For example, compared to the situation in 1991, four times as many people (more than 400,000) needed PEP in 2003. Recent data show that the People's Republic of China, which vaccinates 15 million people per year after potential rabies exposure, spends about US$650 million per year on PEP alone. [ 73 ]
A much more sustainable approach is to prevent the spread of infection at the source, in the animal population, while increasing access to PEP for exposed human patients when needed. Where there is political will and adequate funding to control canine rabies, fatalities can and have been eliminated. Widespread use of dog vaccination has led to the elimination of canine rabies from several countries, including Malaysia in 1954, [ 74 ] Japan in 1956, Taiwan in 1961, Singapore, and, in particular, throughout Western Europe (reviewed in Rupprecht et al, King et al, and Gongal and Wright). [ 75 ]