
All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Alkaptonuria is a congenital enzyme abnormality
Medical expert of the article

Last reviewed: 04.07.2025
One of the very rare metabolic disorders, alkaptonuria, refers to congenital anomalies in the metabolism of the amino acid tyrosine.
This syndrome may also be called homogentisate oxidase deficiency, homogentisinuria, hereditary ochronosis, or black urine disease.[ 1 ]
Epidemiology
According to statistics, there are no more than nine cases of alkaptonuria per 1 million people. And in most European countries, there is one case per 100-250 thousand live births.
Among European countries, the exception is Slovakia (especially the relatively small northwestern region), where the prevalence of alkaptonuria is one case per 19,000 newborns. This is most likely due to the fact that among the Slovak Roma families living there, the level of inbreeding (marriage between cousins) is the highest in Europe: 10-14%. [ 2 ]
Causes alkaptonuria
The exact causes of alkaptonuria, as a congenital disorder of catabolism (metabolic breakdown) of the aromatic (homocyclic) α-amino acid tyrosine, have been established: this type of metabolic disorder is a consequence of homozygous or compound heterozygous mutations of one of the thousands of genes on chromosome 3, more precisely, the HGD gene at locus 3q21-q23 on the long arm of the chromosome. This gene encodes the nucleotide sequences of the liver enzyme homogentisate-1,2-dioxygenase [ 3 ] (also called homogentisic acid oxidase or homogentisate oxidase) - an iron-containing metalloprotein necessary for one of the stages of tyrosine breakdown in the body. [ 4 ], [ 5 ]
Thus, alkaptonuria is a defect of the enzyme homogentisate-1,2-dioxygenase, or more precisely, the result of its genetically determined deficiency or complete absence. [ 6 ]
Being a congenital enzyme deficiency, alkaptonuria is inherited as an autosomal recessive trait, that is, for alkaptonuria to occur in children, both parents must have a modified gene for the enzyme, since each of them passes on to the child only one copy of the gene from the two available.
According to the latest data, there are more than two hundred variants of modification of the HGD gene, and missense mutations, translocation and splicing are most often observed.
Risk factors
The only risk factor for developing this congenital enzymopathy is its presence in the family history and the inheritance of two modified copies of the HGD gene, if the parents do not exhibit alkaptonuria (the risk of transmitting the anomaly is 25%), or one of the parents has this disorder. [ 7 ]
Pathogenesis
Tyrosine plays a key role in the synthesis of proteins, the production of chromoproteins – the skin pigment melanin, as well as thyroid hormones and catecholamine neurotransmitters.
The mechanism for regulating the amount of tyrosine in cells is very complex, and the body normalizes its excess content by breaking it down. The process of tyrosine catabolism, like all aromatic amino acids, is multi-stage and occurs in several stages. Each stage of the metabolic breakdown of tyrosine occurs with the participation of a specific enzyme and the formation of an intermediate compound.
So, first the amino acid is broken down to para-hydroxyphenylpyruvate, which is converted into alkaptone – 2,5-dihydroxyphenylacetic or homogentisic acid. Then the alkaptone should be transformed into maleacetic acid, but this does not happen. [ 8 ]
And the pathogenesis of alkaptonuria consists of the cessation of biochemical reactions of tyrosine catabolism at the stage of formation of homogentisic acid: there is simply no enzyme needed to break it down – homogentisate oxidase.
Homogentisic acid is not used by the body and can accumulate with excretion through the kidneys. In addition, it is oxidized to benzoquinoacetate (benzoquinoneacetic acid), which, by binding to the molecules of tissues and body fluids, forms biopolymer compounds colored like melanin.
The accumulation of these intermediate products in the tissue leads to a disruption of the collagen structure of the cartilage tissue, which reduces its elasticity – with the appearance of many clinical signs of alkaptonuria and the development of complications.
Symptoms alkaptonuria
Alkaptonuria in newborns and infants is characterized by darkening of the urine. When exposed to air, the urine on diapers, nappies, and underwear becomes dark brown; this is due to the accumulation and release of homogentisic acid, which is oxidized to benzoquinoacetate. [ 9 ]
In the absence of other symptoms, alkaptonuria in young children is often not recognized in a timely manner, since urine may darken after several hours of urination. According to some data, only a fifth of children under 12 months who were born with this enzyme deficiency are identified in clinical settings. Therefore, it is very important for parents to pay attention to caring for their infants.
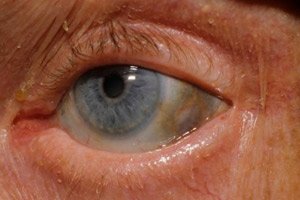
Additionally, early signs include pigmentation (bluish-gray color) of the sclera of the eyes and the cartilages of the ears and nose, which is often called ochronosis.[ 10 ]
Over time, other symptoms appear:
- severe pigmentation of the skin on the cheekbones, armpits and genitals;
- staining of clothing when in contact with sweaty areas of the body;
- attacks of general weakness;
- hoarse voice.
It should be borne in mind that alkaptonuria and ochronosis, as noted above, are synonymous names for the same disorder of tyrosine catabolism.
Maple syrup urine disease and alkaptonuria. Congenital maple syrup urine disease or leucinosis is also a metabolic disorder, has the same inheritance pattern, and even mutations occur on the same chromosome, but affect the gene encoding the enzyme complex of branched-chain α-keto acid dehydrogenase. Because of this, the body cannot break down certain components of proteins, in particular, the amino acids leucine, isoleucine and valine. With this disease, urine (and earwax) have a sweet smell; in addition, the clinical picture of this type of organic acidemia includes hypopigmentation, fluctuations in blood pressure, seizures, vomiting and diarrhea, a drop in blood glucose levels, ketoacidosis, hallucinations, etc. The mortality rate in children is quite high; in adults, without treatment, coma and death may occur due to cerebral edema.
Albinism and alkaptonuria are “united” only by tyrosine. Albinism, including oculocutaneous, is caused by genetic mutations that affect the production of the pigment melanin. Congenital changes are noted in the TYR gene on chromosome 11 (11q14.3), which codes for tyrosinase, a copper-containing melanosome enzyme necessary for the formation of skin pigment based on tyrosine metabolism products. This disease is much more common than alkaptonuria.
Complications and consequences
Caused by the action of intermediate metabolites of tyrosine – homogentisic and benzoquinoneacetic acids – the consequences and complications of alkaptonuria appear due to the deposition of reactive pigmented polymers, the destruction of collagen fibrils and the deterioration of the condition of cartilage (with a decrease in their resistance to mechanical stress).
Over the years, in adulthood, degenerative arthritis and osteoarthritis of large joints (hip, sacroiliac and knee) develop; intervertebral spaces narrow (especially in the lumbar and thoracic spine) – with calcification and formation of osteophytes; the density of the tissue of the subchondral bone plates decreases, and the underlying bones may undergo pathological remodeling with the formation of growths and deformation. [ 11 ]
Damage to the heart valves (aortic and mitral) and coronary arteries may be observed – with signs of coronary heart disease, as well as the formation of stones in the kidneys and prostate gland – due to the same calcification. [ 12 ], [ 13 ]
Diagnostics alkaptonuria
Typically, the diagnosis of congenital metabolic disorders is based on the study of biological fluids of the body.
Based on what tests and reactions can alkaptonuria be diagnosed? Urine tests are required to detect homogentisic acid and determine its level (normal – 20-30 mg per day, elevated – 3-8 g). A urine sample is examined by gas chromatography or mass spectrometry, using liquid chromatography; a screening test for the presence of iron chloride in the urine is possible. [ 14 ]
There is also a method for rapid diagnostics – determining alkapton in dried urine stains on paper (by color intensity).
When clarifying the diagnosis, instrumental diagnostics (radiography) involves identifying radiological signs of osteoarthritis and other joint pathologies in patients.
The diagnosis is confirmed by molecular genetic methods of diagnosing hereditary diseases, such as genetic testing and DNA sequencing. [ 15 ]
Differential diagnosis
Differential diagnosis includes hemochromatosis and acute liver failure of the newborn, melaninuria, acute intermittent porphyria, hemophagocytic lymphohistiocytosis, primary mitochondrial pathology, rheumatoid arthritis, ankylosing spondylitis.
Who to contact?
Treatment alkaptonuria
The main treatment for alkaptonuria is the oral administration of large doses (at least 1000 mg per day) of ascorbic acid. In children, this increases the excretion of homogentisic acid in the urine, and in adults, it reduces the urinary content of its derivative, benzoquinoneacetic acid, and slows its binding to connective tissue structures of the joints and collagen. [ 16 ]
Western European clinics are testing the drug Nitisinone (Orfalin), a drug from the group of metabolites that inhibits the second stage of tyrosine catabolism: the transformation of para-hydroxyphenylpyruvate into homogentisic acid. However, the use of this pharmacological agent leads to the accumulation of tyrosine and can cause severe side effects, including corneal opacity and photophobia, nosebleeds and stomach bleeding, liver failure, changes in the blood, etc. Nevertheless, in the United States, Nitisinone is FDA approved for the treatment of type I tyrosinemia. [17 ], [ 18 ]
As such, physiotherapy treatment – exercise therapy to increase muscle strength and improve joint mobility, balneotherapy and peloid therapy to limit pain – is carried out for joint problems caused by alkaptonuria.
Although tyrosine is not only supplied by food but also produced in the body, patients with alkaptonuria are recommended to follow a low-protein diet and limit the consumption of foods rich in tyrosine, primarily beef and pork, dairy products (especially cheeses), legumes, nuts and seeds.
Prevention
Prevention of gene mutations is impossible, but to prevent the birth of children with a high risk of congenital disorders, there is medical genetic counseling, which is necessary before a planned pregnancy for those couples whose family history includes hereditary diseases. [ 19 ]
Forecast
Fatal outcomes from alkaptonuria are very rare, and death may be caused by serious complications involving the heart and kidneys. So the overall life expectancy of people with alkaptonuria is good.
But the quality of life is reduced due to intense pain in the joints or spine with significant limitation of mobility, often progressive.