Medical expert of the article

New publications
Kidney dysplasia
Last reviewed: 17.10.2021

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
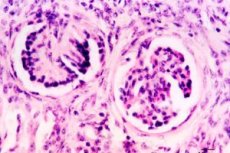
Renal dysplasia occupies a prominent place among the malformations of the urinary system. Renal dysplasia is a heterogeneous group of diseases associated with impaired development of renal tissue. Morphologically, dysplasia is based on impaired differentiation of the nephrogenic blastema and branches of the ureteric sprout, with the presence of embryonic structures in the form of foci of undifferentiated mesenchyme, as well as primitive ducts and tubules. Mesenchyme, represented by polypotent cambial cells and collagen fibers, can form dystontogenetic derivatives of hyaline cartilage and smooth muscle fibers.
Causes of the kidney dysplasia
Pathogenesis
During the morphological study of hypoplastic dysplasia, there is a slight decrease in the mass of the kidneys, there is a lobed surface, division into layers is not always clearly expressed, sometimes there is some expansion or hypoplasia of the ureters. Primitive structures are microscopically detected: many glomeruli are reduced in size, vascular loops are atrophic, the capsule is thickened. The shape of the glomeruli can be S-shaped or ring-shaped, many of them are hyalinized and sclerosed. The glomeruli are located bunches, but are surrounded by loose connective tissue with focal accumulations of lymphoid and histiocytic cells. In the medulla there are many primitive ducts and tubules, which are immature formations of different stages of embryonic development. Primitive ducts, mainly detected in the juxtamedullary zone, are remnants of the mesonephrogenic duct. A characteristic feature is the presence around them of the shadows of smooth muscle cells and connective tissue fibers. The presence of primitive structures reflects the delay in maturation of the nephron.
When morphological study of simple focal dysplasia significant changes in the mass of the kidneys is not observed. In some cases, a decrease in bark thickness is observed. This nephropathy is agnostic on the basis of histological changes detected by microscopy. Simple focal dysplasia, characterized by the presence of clusters of primitive glomeruli and tubules, predominantly in the cortex of the kidneys, surrounded by connective tissue fibers and smooth muscle cells, is sometimes found to be cartilage tissue. The polymorphism of convoluted tubule epithelium is characteristic, where adjacent cells differ in size, configuration, set and number of intracellular organelles. Some children in the kidney may have enlarged tubule lumens. Possible detection and glomerular cysts, but their number is insignificant. In the stroma mononuclear cells of the mesenchymal type are determined.
Simple segmental dysplasia (Ask-Upmark kidney) is quite rare (0.02% of all autopsies). With this type of dysplasia, the kidney is reduced in size, the transverse groove is clearly visible on the outer surface at the site of the hypoplastic segment, and the number of pyramids is reduced. Morphological changes are due to vascular diembriogenesis in individual segments of the kidney, followed by impaired differentiation of tissue structures due to changes in the blood supply to these areas. Usually detected underdevelopment of the corresponding branches of the arteries. A characteristic feature is the presence in the hypoplastic segment of primitive mesonephrogenic ducts, surrounded by smooth muscle cells and foci of hyaline cartilage. In addition, develop sclerosis, glomerular hyalinosis, atrophy of the epithelium of the tubules with the expansion of their lumen, signs of fibrosis and cell infiltration, interstitium.
Aplastic cystic dysplasia (rudimentary multikistosis of the kidney) is 3.5% among all congenital malformations of the urinary system and 19% among all forms of cystic dysplasia Kidneys are significantly reduced in size, constitute a shapeless formation of cysts with a diameter of 2-5 mm, the renal parenchyma is almost completely absent, there is no ureter or there is atresia. Microscopically revealed a large number of cysts, both glomerular and tubular, as well as primitive ducts and foci of cartilage tissue. Bilateral defeat is incompatible with life. Unilateral rudimentary kidney is often detected by random examination, while the second kidney is often abnormal.
Hypoplastic cystic dysplasia (multicystic hypoplastic kidney) is 3.9% among all urinary system defects, and among cystic dysplasia 21.2%. Kidneys are reduced in size and weight. Glomerular cysts are usually located in the subcapsular zone, their diameter is different and can reach 3-5 mm. Tubular cysts are found both in the cortex and in the medulla. Fibrosis of the connective tissue and the presence of primitive ducts are more significant in the medulla. Cysts of large sizes and are cystic extended collecting tubules. The parenchyma of the kidney is partially preserved. Between the pathologically altered areas there are collecting ducts of normal structure. The pelvis can not be changed, more often hypoplastic, like the ureter. Hypoplastic cystic dysplasia is often combined with malformations of the lower urinary tract, gastrointestinal tract, cardiovascular system and other organs.
Bilateral damage early leads to the development of chronic renal failure. As a rule, with the unilateral variant of this dysplasia, the second kidney has some or other manifestations of disembryogenesis.
Hyperplastic cystic dysplasia often accompanies Patau syndrome. The process is two-sided. Kidneys are enlarged, covered with multiple cysts. Microscopy reveals primitive ducts, large cysts in the cortex and medulla. Fatal outcome usually occurs at an early age.
Multicystic dysplasia (multicystic kidney) is a malformation in which the kidneys are enlarged, there are a large number of cysts (5 mm to 5 cm) of various shapes and sizes, between which there is practically no parenchyma.
Microscopic examination between cysts reveals primitive ducts and glomeruli, and areas with cartilage tissue can be found. With a bilateral lesion, death occurs in the first days of life. In case of unilateral lesion, the diagnosis is made randomly by palpation of a tuberous tumor-like formation or by ultrasound. With unilateral multicystic disease, there may be malformations of the second kidney (often hydronephrosis), heart defects, gastrointestinal tract, etc.
In medullary dysplasia (cystic dysplasia of the medullary substance, medullary cystic disease, Fankoni nephronoftiz), the kidneys are usually reduced in size, often embryonic lobulation. The cortical substance is thinned, the medulla is enlarged due to the large number of cysts up to 1 cm in diameter, including the cystic expansion of the collecting tubules. Microscopy shows a decrease in the size of many glomeruli, some of them are hyalinized and sclerosed, the interstitium is also sclerosed, and in the stroma lymphoid infiltration.
Polycystic kidney disease occupies a special place among cystic dysplasia. The emergence of polycystic is associated with a violation of the embryonic development of the kidneys, most often in the form of lack of connection of the primary collecting tubules with a part of the nephron, developing from the metanephrogenic blastoma. The resulting blind tubules continue to develop, they accumulate primary urine, which stretches them, causing epithelial atrophy. At the same time surrounding connective tissue grows.
The size of cysts varies widely: along with small ones, visible only with a magnifying glass or even a microscope, there are large ones, up to several centimeters in diameter. A large number of thin-walled cysts in the cortex and the medulla of the kidneys attached to them in the section of the form of honeycombs. Histologically, cysts are represented by dilated tubules with a cubic epithelium or have the appearance of cavities with a thick connective tissue wall and sharply flattened epithelium. E. Potter (1971) described cysts associated with the expansion of the bowmen capsule of the glomeruli, without changing the tubules. Cysts can be empty or contain serous, protein liquid, sometimes painted with blood pigments, uric acid crystals. Kidney stroma in polycystic sclerosis, often with focal lymphoid cell infiltration, and in children under 1 year old - with foci of extramedullary hematopoiesis. Sometimes cartilage islets or smooth muscle fibers are detected in the stroma. The number and type of glomeruli and tubules located between the cysts may be different
Symptoms of the kidney dysplasia
Simple total dysplasia in the literature is often described as hypoplastic dysplasia. Among all the congenital malformations of the urinary system, it is 2.7%.
There are aplastic and hypoplastic variants. In the aplastic variant of renal dysplasia in the case of bilateral lesions, death occurs in the first hours or days of life.
The hypoplastic variant is characterized by an early manifestation of urinary syndrome characterized by mosaicism and the early development of chronic renal failure.
Simple focal dysplasia is diagnosed, as a rule, during nephrobiopsy or autopsy. Clinical manifestations of the disease are absent.
In simple segmental dysplasia, the dominant symptom is the development of resistant arterial hypertension already at an early age, which is more common in girls. Children complain of a headache, there may be convulsions, and early changes in the fundus vessels develop.
One of the main clinical symptoms is pain in the form of abdominal pain, polyuria and polydipsia appear quite early as manifestations of tubulo-interstitial syndrome. In some cases, there is a lag in body weight and child growth. Urinary syndrome is manifested predominant proteinuria on the background of microhematuria and moderate leukocyturia.
Clinical signs of polycystic kidney disease appear in adolescence: back pain, palpation of the tumor-like formation in the abdominal cavity, arterial hypertension. Urinary syndrome is manifested by hematuria. Often joins pyelonephritis. Functionally, the kidneys are preserved for many years, then hyposthenuria, reduction of glomerular filtration and azotemia appear.
A multilocular cyst (focal cystic dysplasia of the kidneys) is a focal form of cystic dysplasia of the kidney and is characterized by the presence of a multi-chamber cyst at one of its poles, bounded by a capsule from normal kidney tissue and divided inside by partitions.
The clinical picture of a multilocular cyst is characterized by the appearance of pain syndrome of varying severity in the abdomen and in the lumbar region due to impaired urine outflow due to compression of a large cyst of the pelvis or ureter. In addition, in connection with the possible compression of the abdominal organs, symptoms appear that simulate their disease.
Clinical manifestations of medullary dysplasia usually develop after reaching the age of 3 years; more often, at the age of 5-6 years old, Fankoni symptom complex appears - polyuria, polydipsia, fever, lag in physical development, repeated vomiting, dehydration, acidosis, anemia, rapid progression uremia.
The clinical picture of aplastic cystic dysplasia is determined by the state of the second kidney, in which pyelonephritis often develops due to the presence of dysplasia in it.
Multicystic dysplasia can be manifested by the presence of dull or paroxysmal abdominal pain, as well as in the lumbar region. Possible detection of arterial hypertension.
In cortical dysplasia (renal microcystic disease, congenital nephrotic syndrome of the "Finnish" type), the kidneys are not changed in size, the lobulation can be maintained. Small glomerular and tubular cysts with a diameter of 2-3 mm are found. A picture of nephrotic syndrome has been observed since birth. Congenital nephrotic syndrome of the "Finnish" type is hormone resistant, with a poor prognosis. Early development of chronic renal failure is noted.
The clinical picture of hypoplastic cystic dysplasia is due to pyelonephritis, the development of chronic renal failure, the rate of progression of which depends not only on the number of intact hypoplassed kidney parenchyma, but also on the extent of damage to the second non-hypoplastic kidney, but usually with dysplastic elements.
Hypoplastic dysplasia can be detected on the background of an intercurrent disease, while extrarenal syndromes may be absent or weakly expressed. Urinary syndrome is manifested by hematuria with moderate proteinuria. Manifestations of this disease are very heterogeneous. Often there can be a proteinuric variant with significant loss of protein, but edematous syndrome is relatively rare, even with significant proteinuria, and the nephrotic syndrome is characterized as incomplete. Dynamic observation of the child shows that the clinical picture is subsequently characterized by nephrotic syndrome, the presence of tubulo-interstitial changes, often with a layering of urinary tract infection.
It is characteristic of children with hypoplastic dysplasia to develop hypoimmune or immunodeficient states, which explains the addition of severe and frequent intercurrent diseases with the progression of the pathological process in the kidneys. An important feature of this nephropathy is the absence of high blood pressure, hypotension is more common. Increased blood pressure occurs already with the development of chronic renal failure.
The course of hypoplastic dysplasia is torpid, there is no cyclicity or wave-like character of the manifestations, and drug therapy is usually ineffective.
Forms
Currently, there is no generally accepted classification of kidney dysplasia. Most of the authors, based on the morphological manifestations, distinguish between simple and cystic dysplasia, by localization - cortical, medullary, cortico-medullary. Depending on the prevalence, focal, segmental, total dysplasias are distinguished.
Depending on the prevalence, total, focal and segmental forms of cystic dysplasia are distinguished.
Aplastic, hypoplastic, hyperplastic and multicystic variants are distinguished from total forms of cystic dysplasia.
Polycystic is manifested in two main forms, which differ in the pattern of inheritance, clinical manifestations, morphological picture - the "infantile" and "adult" types.
Polycystic "infantile" type (small cystic kidney) has an autosomal recessive mode of inheritance. The kidneys are significantly enlarged in size and mass. In the cortex and medulla numerous cysts are cylindrical and spindle shaped. Cysts are delimited by scant layers of connective tissue. Cysts are also found in the liver and other organs. Clinical manifestations depend on the number of affected tubules. With the defeat of up to 60% of the tubules, death from progressive uremia occurs in the first 6 months. The results of OV Chumakova (1999) do not confirm the classical ideas about the early mortality of children with an autosomal recessive form of polycystosis and show that their lifespan can be quite long, even with early detection of clinical symptoms. However, chronic renal failure in them develops earlier than in the autosomal dominant form of polycystic disease. In these patients, symptoms of liver damage play a leading role in the clinical picture. The clinic is often marked micro-, gross hematuria and increased blood pressure. Polycystic often complicated by torpid pyelonephritis.
In case of polycystic "adult" type (large cystic kidney), the kidneys are almost always enlarged, their weight in adults reaches up to 1.5 kg and more each. In the cortex and medulla there are numerous cysts up to 4-5 cm in diameter.
Diagnostics of the kidney dysplasia
Diagnosis of polycystic kidney disease is based on a family history, ultrasound data, excretory urography, in which there is an increase in the contours of the kidneys, flattening of the pelvis with elongation, lengthening and compression of the calyxes.
In the diagnosis of multilocular cyst, radiological methods of examination, including nephrotography and angiography, are of crucial importance.
Among the laboratory signs of medullary dysplasia, hypoproteinemia is characteristic, and urinary syndrome usually manifests with a small proteinuria. In connection with the increased loss of salts, hyponatremia, hypokalemia and hypocalcemia develop. Acidosis develops due to significant bicarbonaturia, impaired acido-and ammoniogenesis.
Diagnosis of aplastic cystic dysplasia is based on ultrasound data, excretory urography, reno-and scintigraphy. When cystoscopy, the mouth of the ureter on the side of the rudimentary kidney, as a rule, is absent or stenotic.
For the diagnosis of hypoplastic dysplasia, random detection of the disease, the presence of multiple stigmas of disembryogenesis, and a slight delay in physical development are important.
What tests are needed?
Differential diagnosis
Treatment of the kidney dysplasia
Symptomatic treatment of hypoplastic dysplasia.
In case of detection of multicystosis, nephrectomy is performed at the risk of the development of malignancy.
Treatment of medullary dysplasia is symptomatic. With the development of chronic renal failure, hemodialysis or peritoneal dialysis and kidney transplantation is indicated.
Forecast
The prognosis for hypoplastic dysplasia is serious, with early development of chronic renal failure and the need to organize replacement therapy - hemodialysis or peritoneal dialysis, kidney transplantation.
 [38]
[38]