Medical expert of the article

New publications
Cystic Fibrosis
Last reviewed: 23.04.2024

All iLive content is medically reviewed or fact checked to ensure as much factual accuracy as possible.
We have strict sourcing guidelines and only link to reputable media sites, academic research institutions and, whenever possible, medically peer reviewed studies. Note that the numbers in parentheses ([1], [2], etc.) are clickable links to these studies.
If you feel that any of our content is inaccurate, out-of-date, or otherwise questionable, please select it and press Ctrl + Enter.
Cystic fibrosis is a genetic autosomal recessive monogenic disease characterized by impaired secretion of the exocrine glands of vital organs with the defeat primarily of the respiratory and digestive systems, severe course and unfavorable prognosis.
 [1]
[1]
Epidemiology
The incidence of cystic fibrosis varies from 1: 2,500 to 1: 4,600 newborns. Annually in the world about 45000 patients with cystic fibrosis are born. The frequency of carriers of the gene of cystic fibrosis is 3-4%, there are about 275 million people on the globe, carriers of this gene, about 5 million of them live in Russia, about 12.5 million live in the countries of the NF.
Causes of the cystic fibrosis
Cystic fibrosis is transmitted by an autosomal recessive type. The gene of cystic fibrosis is located in the 7 autosome, contains 27 exons and consists of 250,000 pairs of nucleotides.
In one gene, many mutations are possible, each of which is characteristic of a particular population or geographic region. More than 520 mutations are described, the most common of them is delta-P-508, i.e. Replacement of the amino acid phenylalanine in 508 positions.
Pathogenesis
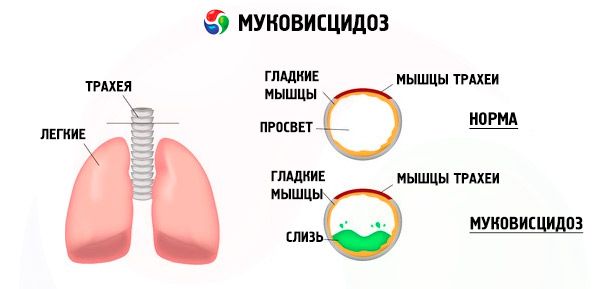
Due to mutations of the cystic fibrosis gene, the structure and function of the protein called the CFTR-cystic fibrosis transmembrane regulator are disrupted. This protein acts as a chloride channel involved in the water-electrolyte metabolism of epithelial cells of the bronchopulmonary system, the gastrointestinal tract, the pancreas, the liver, and the reproductive system. As a result of the violation of the function and structure of the CFTR protein, chlorine ions accumulate inside the cell. Cl - ). This leads to a change in the electrical potential in the lumen of the excretory ducts, which contributes to the intake of a large number of sodium ions (Na + ) from the lumen of the duct into the cell and then to the enhanced absorption of water from the cell space.
As a result of these changes, the secret of most of the glands of external secretion thickens, its evacuation is violated, which leads to pronounced secondary disorders in organs and systems, most pronounced in the bronchopulmonary and digestive systems.
In bronchi, a chronic inflammatory process of varying intensity develops, the function of the ciliated epithelium develops, sputum becomes very viscous, thick, very difficult to evacuate, its stagnation is observed, bronchiolo- and bronchiectasis are formed, which eventually become more common. These changes lead to an increase in hypoxia and the formation of a chronic pulmonary heart.
Patients with cystic fibrosis are extremely predisposed to the development of a chronic inflammatory process in the bronchopulmonary system. This is due to pronounced impairments in the system of local bronchopulmonary protection (lowering the level of IgA, interferon, phagocytic function of alveolar macrophages and leukocytes).
An important role in the development of chronic inflammation in the bronchopulmonary system belongs to alveolar macrophages. They produce a large number of IL-8, which dramatically increases the chemotaxis of neutrophils in the bronchial tree. Neutrophils accumulate in large quantities in the bronchi and together with epithelial cells release a lot of anti-inflammatory cytokines, including IL-1, 8, 6, tumor necrosis factor, and leukotrienes.
An important role in the pathogenesis of the lesion of the bronchopulmonary system is also played by the high activity of the enzyme elastase. There are exogenous and endogenous elastase. The first is produced by the bacterial flora (especially the Pseudomonas aeruginosa), the second by neutrophilic leukocytes. Elastase destroys the epithelium and other structural elements of the bronchi, which contributes to the further disruption of mucociliary transport and the fastest formation of bronchiectasias.
Neutrophilic leukocytes also excrete other proteolytic enzymes. They counteract the effect of proteolytic enzymes and, consequently, protect the bronchopulmonary system from their damaging effect of a1-anpripinsin and the secretory inhibitor of leukoproteases. However, unfortunately, in patients with cystic fibrosis, these protective factors are suppressed by a significant amount of neutrophil protease.
All these circumstances contribute to the introduction of infection in the bronchopulmonary system, the development of chronic purulent bronchitis. In addition, it should be taken into account that the defective protein encoded by the cystic fibrosis gene changes the functional state of the bronchial epithelium, which favors adhesion to the bronchial epithelium of bacteria, primarily the Pseudomonas aeruginosa.
Along with the pathology of the bronchopulmonary system in cystic fibrosis, there is also a marked defeat of the pancreas, stomach, thick and small intestine, and liver.
Symptoms of the cystic fibrosis
Cystic fibrosis manifests itself in a variety of clinical symptoms. In newborns, the disease can manifest with a meconial ileus. Due to the lack or even complete absence of trypsin, the meconium becomes very dense, viscous, accumulates in the ileocecal region. Further, intestinal obstruction develops, which is manifested by intense vomiting with an admixture of bile, bloating, lack of meconium secretion, development of peritonitis symptoms, rapid growth of clinical manifestations of severe intoxication syndrome. A child may die in the first days of life unless urgent surgical intervention is performed.
In less severe cases, a characteristic sign of cystic fibrosis is an abundant, frequent stool, ointment, with a lot of fat, with a very unpleasant odor. In 1/3, patients there is a prolapse of the rectum.
Subsequently, intestinal dysfunction persists in patients, malabsorption syndrome develops, severe disruption of physical development, severe hypovitaminosis.
In the first or second year of the child's life, symptoms of a bronchopulmonary system (mild form of the disease) appear, which is manifested by a cough that can be extremely pronounced and resemble a cough in whooping cough. Cough accompanied by cyanosis, shortness of breath, separation of thick sputum, initially mucous, and then purulent. Gradually a clinical picture of chronic obstructive bronchitis and bronchiectasis, emphysema of the lungs and respiratory failure is formed. Children are extremely susceptible to acute respiratory-viral and bacterial infections, which contributes to exacerbations and progression of bronchopulmonary pathology. Possible the development of infectious-dependent bronchial asthma.
In children of school age, cystic fibrosis may manifest as "intestinal colic." Patients complain of severe paroxysmal pains in the abdomen, bloating, repeated vomiting. When palpating the abdomen, dense formations are identified, located in the projection of the large intestine - feces, mixed with thick dense mucus. Children are very predisposed to the development of hypochloraemic alkalosis due to excessive salt removal with sweat in hot weather, while "salt frost" appears on the skin of the child.
The defeat of the bronchopulmonary system in adults
The defeat of the bronchopulmonary system in patients with cystic fibrosis (pulmonary form of the disease) is characterized by the development of chronic purulent obstructive bronchitis, bronchiectasis, chronic pneumonia, emphysema, respiratory failure, pulmonary heart. Some patients develop pneumothorax and other complications of cystic fibrosis: atelectasis, lung abscesses, hemoptysis, pulmonary hemorrhage, infectious-dependent bronchial asthma.
Patients complain of a painful paroxysmal cough with a very viscous, difficult to separate mucopurulent sputum, sometimes with an admixture of blood. In addition, dyspnoea is very characteristic at first under physical stress, and then at rest. Dyspnea is due to bronchial obstruction. Many patients complain of chronic rhinitis caused by polyposis and sinuititis. There is also a significant weakness, a progressive decline in performance, frequent acute respiratory-viral diseases. On examination, attention is paid to the pallor of the skin, puffiness of the face, cyanosis of the visible mucous membranes, pronounced dyspnea. With the development of a decompensated pulmonary heart, edema on the legs appears. There may be thickening of the terminal phalanges of the fingers of the hands in the form of tympanic sticks, and nails - in the form of hourglasses. The thorax acquires a barrel shape (due to the development of emphysema).
When percussion of the lungs are determined signs of emphysema - box sound, a sharp restriction of the mobility of the pulmonary edge, the lowering of the lower border of the lungs. With auscultation of the lungs, hard breathing with prolonged exhalation is revealed, scattered dry wheezes, moist medium and small bubbling rales. With a pronounced emphysema of the lungs, the breathing is sharply weakened.
Extrathoracic manifestations of cystic fibrosis
Extrapulmonary manifestations of cystic fibrosis can be quite pronounced and occur frequently.
Defeat of the pancreas
Insufficiency of the exocrine function of the pancreas of varying degrees is observed in 85% of patients with cystic fibrosis. With minor pancreatic damage, there are no maldigestion and malabsorption syndromes, only laboratory manifestations of exocrine insufficiency (low level of trypsin and lipase in the blood and duodenal contents, often expressed steatorrhoea). It is known that in order to prevent the syndrome of maldigestion, only 1 to 2% of the total lipase is secreted. Clinically, only significant violations of the external secretory function are manifested.
Under normal conditions, the secretion of a liquid consistency rich in enzymes is produced in the acini of the pancreas. With the progress of the secretion through the excretory duct, it is enriched with water and anions, and it becomes even more liquid. In cystic fibrosis, due to a violation of the structure and function of the transmembrane regulator (chloride channel), a sufficient amount of liquid does not enter the secret of the pancreas, it becomes viscous, and the rate of its progress along the excretory duct slows down dramatically. Secret proteins are deposited on the walls of small excretory ducts, as a result of which their obstruction develops. As the disease progresses, eventually destruction and atrophy of the acini develop - chronic pancreatitis with exocrine pancreatic insufficiency is formed. This is clinically reflected in the development of syndromes of maldigestia and malabsorption. Pancreatic insufficiency is the main cause of malabsorption of fat in cystic fibrosis, however, as a rule, this is observed with a significant lipase deficiency. Forsher and Durie (1991) indicate that in the absence of pancreatic lipase, fat is split and absorbed by 50-60%, due to the presence of gastric and salivary (sublingual) lipases, whose activity is close to the lower limit of the norm. Along with the violation of the splitting and absorption of fats, there is a violation of the cleavage and reabsorption of proteins. With feces, about 50% of the protein fed with food is lost. Absorption of carbohydrates suffers less in spite of the deficiency of α-amylase, however, carbohydrate metabolism can be significantly impaired.
The defeat of the pancreas is expressed in the development of the syndrome of maldigestia and malabsorption with significant weight loss, abundant fatty stool.
Development of syndromes of maldigestia and malabsorption is also facilitated by severe impairment of the function of the intestinal glands, a violation of the secretion of intestinal juice and a decrease in the content of intestinal enzymes in it.
Syndromes of maldigestia and malabsorption are also called the intestinal form of cystic fibrosis.
Violation of the incremental function of the pancreas (diabetes mellitus) is observed in patients with cystic fibrosis in the late stages of the disease (in 2% of children and in 15% of adults)
Lesion of the liver and bile ducts
In 13% of patients with mixed and intestinal cystic fibrosis, cirrhosis develops. It is most typical for mutations of W128X, delta-P508 and X1303K. In 5-10% of patients, biliary cirrhosis with portal hypertension is found. According to Welch, Smith (1995), clinical, morphological, laboratory, instrumental signs of liver damage are found in 86% of patients with cystic fibrosis.
Many patients with cystic fibrosis also develop chronic cholecystitis, often calculous.
Violation of the function of the sexual glands
In patients with cystic fibrosis, azoospermia can occur, which is the cause of infertility. Reduced fertility is also characteristic of women.
Stages
There are three degrees of severity of the pulmonary form of cystic fibrosis.
The mild form of cystic fibrosis is characterized by rare exacerbations (not more often than once a year), during periods of remission clinical manifestations are practically absent, the sick are able to work.
The course of moderate severity - exacerbation is observed 2-3 times a year and lasts about 2 months and longer. In the acute phase, intense cough with hard-to-separate sputum is observed, shortness of breath even with insignificant physical exertion, subfebrile body temperature, general weakness, sweating. At the same time there is a violation of the exocrine function of the pancreas. In the phase of remission, working capacity is not completely restored, dyspnoea is retained during physical exertion.
Severe course is characterized by very frequent exacerbations of the disease. There are practically no remissions. In the clinical picture, severe respiratory failure, symptomatology of the chronic pulmonary heart, often decompensated, characterized by hemoptysis, is at the forefront. There is a significant decrease in body weight, the patients are completely disabled. As a rule, severe bronchopulmonary pathology is accompanied by a pronounced impairment of the pancreas function.
Forms
- Broncho-pulmonary lesions
- Repeated and recurrent pneumonia with prolonged course.
- Abscessed pneumonia, especially in infants.
- Chronic pneumonia, especially bilateral.
- Bronchial asthma, refractory to conventional therapy.
- Recurrent bronchitis, bronchiolitis, especially with the seeding of Pseudomonas aeruginosa.
- Changes in the gastrointestinal tract
- Meconial ileus and its equivalents.
- Syndrome of impaired intestinal absorption of unknown origin.
- Jaundice obstructive type in newborns with protracted course.
- Cirrhosis of the liver.
- Diabetes.
- Gastroesophageal reflux.
- Chololithiasis.
- Prolapse of the rectum.
- Changes in other organs and systems
- Disorders of growth and development.
- Delayed sexual development.
- Male infertility.
- Polyps of the nose.
- Sibs from families with patients with cystic fibrosis.
[24]
Complications and consequences
Complications from the gastrointestinal tract include:
- Diabetes mellitus develops in 8-12% of patients over the age of 25.
- Fibrotic colonopathy.
- Meconial obstruction of the intestine in the neonatal period (in 12% of newborns with cystic fibrosis, distal syndrome of intestinal obstruction, rectal prolapse, peptic ulcer and gastroesophageal reflux disease.
Complications from the liver:
- Fatty degeneration of the liver (in 30-60% of patients),
- Focal biliary cirrhosis, multinodular biliary cirrhosis, and associated portal hypertension.
Portal hypertension sometimes leads to death due to varicose veins of the esophagus.
The prevalence of cholecystitis and cholelithiasis is higher in patients with cystic fibrosis than in other individuals.
Delayed puberty and decreased fertility and other complications. Most men have azoospermia and underdevelopment of the vas deferens.
Diagnostics of the cystic fibrosis
A common blood test is characterized by anemia of varying severity, usually normo- or hypochromic. Anemia has a multifactorial genesis (decrease in absorption of iron and vitamin B12 in the intestine due to the development of malabsorption syndrome). Possible leukopenia, with the development of purulent bronchitis and pneumonia - leukocytosis, an increase in ESR.
The general analysis of urine - without significant changes, in rare cases there is insignificant proteinuria.
Coprologic examination - there is steatorrhoea, creatorrhea. Becker (1987) recommends measurement in the feces of chymotrypsin and fatty acids. Before the determination of chymotrypsin in the stool, it is necessary to cancel the intake of digestive enzymes not less than 3 days before the test. In cystic fibrosis, the amount of chymotrypsin in feces is reduced, and the number of fatty acids is increased (the normal release of fatty acids is less than 20 mmol / day). It should be taken into account that increased excretion with feces of fatty acids is also observed when:
- deficiency of conjugated fatty acids in the small intestine with hepatic insufficiency, biliary tract obstruction, significant bacterial colonization of the small intestine (intensive hydrolysis of bile acids occurs);
- ileite;
- celiac disease (with the development of malabsorption syndrome);
- enteritis;
- intestinal lymphomas;
- Whipple's disease;
- food allergies;
- accelerated transit of food masses for diarrhea of different genesis, carcinoid syndrome, thyrotoxicosis.
The biochemical analysis of blood reduces the content of total protein and albumin, increases the level of alpha2 and gamma globulins, bilirubin and aminotransferases (with liver damage), reduces the activity of amylase, lipase, trypsin, and iron, calcium (with the development of maldigestia syndrome, malabsorption).
Sputum analysis - the presence of a large number of neutrophilic leukocytes and microorganisms (with sputum smear).
Investigation of the absorption function of the small intestine and the exocrine function of the pancreas - significant violations are detected.
X-ray examination of the lungs - reveals changes, the severity of which depends on the severity and phase of the disease. The most characteristic changes are:
- increase in the intensity of the pulmonary pattern due to peribronchial interstitial changes;
- expansion of the roots of the lungs;
- pattern of lobular, subsegmental or even segmental atelectasis of the lungs;
- increased transparency of pulmonary fields, mainly in the upper parts, low standing and insufficient diaphragm mobility, widening of the vaginal space (manifestation of emphysema of the lungs);
- segmental or polysegmentary infiltration of pulmonary tissue (with the development of pneumonia).
Bronchography - reveals the changes caused by obstruction of the bronchi by viscous sputum (fragmentation of bronchial filling with contrast, uneven contours, the phenomenon of bronchus rupture, a significant decrease in the number of lateral branches), and bronchoelectrodes (cylindrical, mixed) localized mainly in the lower parts of the lungs.
Bronchoscopy - detects a diffuse purulent bronchitis with an abundant amount of thick, viscous sputum and fibrinous films.
Spirography - already in the early stages of the disease, reveals respiratory failure of the obstructive type (decrease in FVC, FEV1 of the Tiffno index), restrictive (decrease in ZHEL) or most often obstructive-restrictive (decrease in ZHEL, FVC, FEV1, Tiffno index).
A sweat test (a study of sweat electrolytes) by Gibson and Cook - stimulation of sweating with the help of electrophoresis with pilocarpine followed by determination in the sweat chloride. Doerehuk (1987) describes the sample as follows. Electrophoresis pilocarpine is produced in the forearm area, the electric current is 3 mA. After cleansing the skin with distilled water, sweat is collected with filter paper applied to the stimulated area, covered with gauze to avoid evaporation from it. After 30-60 minutes, the filter paper is removed and eluted in distilled water. Measure the amount of collected sweat. To obtain reliable results, it is necessary to collect at least 50 mg (preferably 100 mg) of sweat.
With a chloride concentration of more than 60 mmol / l, the diagnosis of cystic fibrosis is considered probable; at a chloride concentration of more than 100 mmol / l - reliable; while the difference in the concentration of chlorine and sodium should not exceed 8-10 mmol / l. Hadson (1983) recommends a sample with prednisolone at the boundary value of sodium and chloride contents (taking 5 mg orally for 2 days, followed by electrolytes in sweat). In persons who do not have cystic fibrosis, the sodium level in the sweat decreases to the value of the lower limit of the norm, with cystic fibrosis - it does not change. A sweating sample is recommended for every child with a chronic cough.
The analysis of blood spots or DNA samples on major mutations of the cystic fibrosis gene is the most sensitive and specific diagnostic test. However, this method is suitable for countries where the mutation frequency of delta-P508 is above 80%. In addition, the technique is very expensive and technically complex.
Prenatal diagnosis of cystic fibrosis - is made by determining the isoenzymes of alkaline phosphatase in the amniotic fluid. This method is possible with 18-20 weeks of pregnancy.
The main criteria for diagnosis of cystic fibrosis are the following:
- indications in the history of childhood retardation in physical development, recurrent chronic respiratory diseases, dyspeptic disorders and diarrhea, presence of cystic fibrosis in close relatives;
- chronic obstructive bronchitis, often recurrent, with the development of bronchiectasis and emphysema, often recurrent pneumonia;
- chronic recurrent pancreatitis with marked decrease in exocrine function, malabsorption syndrome;
- increased chlorine content in the sweat of the patient;
- infertility with preserved sexual function.
Successful diagnosis and differential diagnosis of cystic fibrosis is facilitated by the identification of risk groups.
The program of examination for cystic fibrosis
- General analysis of blood, urine, sputum.
- Bacteriological analysis of sputum.
- Coprologic analysis.
- Biochemical analysis of blood: determination of the content of total protein and protein fractions, glucose, bilirubin, aminotransferases, alkaline phosphatase, gamma-glutamyltranspeptidases, potassium, calcium, iron, lipase, amylase, trypsin.
- Examination of the exocrine function of the pancreas and the intestinal absorption function.
- X-ray and lung radiography, lung CT.
- ECG.
- Echocardiography.
- Bronchoscopy and bronchography.
- Spirography.
- A sweat test.
- Consultation of a geneticist.
- Analysis of blood spots or DNA samples for major mutations of the cystic fibrosis gene.
What tests are needed?
Who to contact?
Treatment of the cystic fibrosis
The type and severity of the symptoms of cystic fibrosis can be very different, so there is no typical treatment plan, it is individual in each case.
Therapy consists of the following treatment measures:
- Breathing exercises and postural drainage, helping to get rid of the thick mucus that accumulates in the lungs. Some methods of airway cleansing require help from family members, friends or a pulmonologist. Many people use an inflatable breast vest that vibrates with a high frequency.

- Inhalation drugs that exert bronchodilator, draining (mucolytics) and antibacterial effects (eg, fluoroquinolones).
- Preparations containing pancreatic enzymes to improve digestion. These drugs are taken with food.
- Multivitamins (including fat-soluble vitamins).
In 2015, the FDA approved a second drug for the treatment of cystic fibrosis, which affects a defective protein known as CFTR. The first drug, the so-called CFTR modulator, was approved in 2012. It is expected that CFTR modulators can prolong the life of some people with cystic fibrosis for dozens of years.
Surgical treatment may be required to treat the following respiratory complications:
- Pneumothorax, massive recurrent or persistent hemoptysis, nasal polyps, persistent and chronic sinusitis.
- Meconial obstruction, intussusception, prolapse of rectum.
Lung transplantation is performed at the terminal stage of the disease.
Forecast
The average age of survival of patients with cystic fibrosis varies from 35 to 40 years. The average age of survival in men is higher than that of women.
Thanks to modern treatment strategies, 80% of patients reach adulthood. Nevertheless, cystic fibrosis significantly limits the functional capabilities of the patient. The medicine for this disease has not yet been developed.